La hemostasia es el mecanismo de defensa del organismo que conduce al cese del sangrado de un vaso sanguíneo tras un traumatismo o lesión. El proceso implica múltiples pasos interrelacionados, que culminan en la formación de un «tapón» o coágulo que sella el área dañada del vaso sanguíneo para controlar el sangrado mientras se lleva a cabo el proceso de regeneración de un tejido. Una vez que la lesión comienza a sanar, el «tapón» se remodela y disuelve lentamente con la restauración del tejido normal en el sitio dañado. No obstante, existen trastornos de coagulación sanguínea que afectan a este mecanismo de defensa. A continuación, hablamos de algunos de ellos y ofrecemos las opciones que propone el fabricante BioMedica Diagnostics para su determinación y diagnosis.
Síndrome Antifosfolípido
El síndrome antifosfolípido es una enfermedad autoinmune que se caracteriza por un estado de hipercoagulabilidad debido a la presencia de anticuerpos fosfolípidos en la sangre. Estos anticuerpos antifosfolípidos afectan al proceso normal de coagulación, aumentando la formación de coágulos al interaccionar con proteínas implicadas las diferentes rutas de coagulación:
- Inhibir la proteína C (proteína reguladora en la vía común de coagulación).
- Aumentan la formación de protrombina.
- Disminuir la activación de la proteína C al afectar a su cofactor la proteína S.
Los anticuerpos antifosfolípidos son anticuerpos cuya diana son los fosfolípidos de las membranas celulares. En concreto, se dividen en dos grupos:
- Anticuerpos anticardiolipinas: dirigidos contra la cardiolipina, componente de las membranas mitocondriales.
- Anticuerpos anticoagulantes lúpico: dirigidos contra los complejos fosfolípido-proteína. En este grupo destacan los anticuerpos anti-β2 glicoproteína I.
El síndrome antifosfolípido provoca coágulos en venas y/o arterias. En el caso de mujeres embarazadas se relaciona con complicaciones tales como abortos espontáneos tempranos o tardíos, muerte fetal, partos pretérminos y preeclampsia severa.
El diagnóstico del síndrome antifosfolípido consiste en la aparición de un evento clínico tal como coágulos sanguíneos recurrentes y en el caso de las mujeres, abortos espontáneos; seguidos de dos análisis para la detección de estos anticuerpos antifosfolípidos en sangre, espaciados al menos tres meses.
En BioMedica Diagnostics encontrarás los siguientes kits para la determinación del síndrome antifosfolípido:
- DVVtest, referencias 810 y 825: para la determinación de anticoagulantes lúpicos en plasma humano.
- DVV confirm, referencias 815 y 815L: reactivo de coagulación con alto contenido de fosfolípidos.
- ACTICLOT dPT Dilute Prothrombin Time Test, referencia 824: test de tiempo de protrombina diluida completamente integrada para detectar y confirma la presencia de anticuerpos fosfolípido-dependientes.
- LAtrol LA Control Plasmas, referencias 816A y 816N: plasmas de control desarrollados para su uso como parte de los procedimientos diarios de control de calidad para las pruebas de anticoagulante de lupus.
Puedes encontrar más información en nuestro post anterior: Síndrome Antifosfolípido: kits de diagnóstico para la detección de anticoagulantes lúpicos.
Púrpura Trombocitopénica Trombótica
La púrpura trombocitopénica trombótica es una enfermedad hemorrágica microangiopática (rotura de los glóbulos rojos durante su circulación por la sangre) que se caracteriza por la presencia de moretones en la piel (de ahí el uso de púrpura en el nombre), disminución del número de plaquetas (trombocitopenia) y la formación de trombos (trombótica) en pequeños vasos sanguíneos.
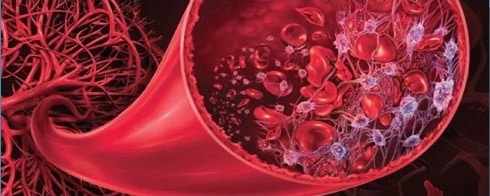
En la púrpura trombocitopénica trombótica (i) las plaquetas se agregan de forma espontánea y (ii) la cascada de coagulación se activa en pequeños vasos vasculares. Ambos procesos desembocan en la formación de microtrombos en el torrente sanguíneo, los cuales causan la rotura de glóbulos rojos a su paso.
La púrpura trombocitopénica trombótica puede manifestarse de dos formas: idiopática o secundaria. La púrpura trombocitopénica trombótica idiopática es hereditaria y se debe a un déficit de la enzima ADAMTS13. Esta enzima, metaloproteinasa, rompe el factor de von Willebrand, lo que permite que este factor interaccione con las plaquetas induciendo la agregación plaquetaria y por tanto la formación de microtrombos intravasculares. Esto conlleva que haya menos plaquetas disponibles en la sangre y de ahí que los pacientes presenten un menor número de recuento plaquetario.
El origen de la púrpura trombocitopénica trombótica secundaria aún no está establecido, pero se cree que en estos casos la hemorragia microangiopática pueda derivar de daños causados a nivel endotelial producidos por: cáncer, quimioterapia, trasplante de células madre hematopoyéticas, infección por VIH, terapia de reemplazo hormonal y estrógenos y medicamentos tales como ticlopidina, clopidogrel, quinina y ciclosporina A.
En BioMedica Diagnostics encontrarás los siguientes kits para la determinación de la púrpura trombocitopénica trombótica:
- Actifluor ADAMTS13 Activity Assay, referencia 812: ensayo de transferencia de energía por resonancia de fluorescencia (FRET) para la medición de ADAMTS13 en plasma humano.
- IMUBIND ADAMTS13 ELISA, referencia 813: diseñado para la medición de anticuerpos ADAMTS13 IgG en plasma humano.
- IMUBIND ADAMTS13 Autoantibody ELISA, referencia 814: diseñado para la medición de anticuerpos ADAMTS13 IgG en plasma humano.
Si estás interesado en este tema, puedes encontrar más información en nuestro post anterior: Diagnóstico de Púrpura Trombótica Trombocitopénica (PTT).
Von Willebrand
La enfermedad de von Willebrand es consecuencia de bajos niveles del factor von Willebrand, el cual es necesario para la adhesión y agregación plaquetaria. Por lo que la deficiencia de este factor impide la formación del coágulo.
Hay cuatro tipos de la enfermedad y aunque sea una enfermedad hereditaria se han dado formas adquiridas.
Von Willebrand es la anomalía en la coagulación de carácter hereditario más común entre la población. Aún así, su prevalencia es aproximadamente de una de cada cien personas. Sin embargo, la mayoría de los pacientes no presenta síntomas, solo uno de cada diez mil afectados representa un caso clínicamente significativo.
BioMedica Diagnostics dispone de los siguientes kits para la determinación de la enfermedad de von Willebrand:
- IMUBIND vWF Activity ELISA, referencia 885. Ensayo cuantitativo directo de inmunoabsorción enzimática para la detección del factor von Willebrand en plasma humano citratado.
- IMUBIND vWF ELISA, referencia 828. Ensayo por inmunoabsorción ligado a enzimas para la medición del antígeno FvW en plasma humano.
Puedes encontrar más información en nuestro post anterior: Kits ELISA para detección de la enfermedad de von Willebrand.
REFERENCIAS
Autor: Gago Fuentes, Raquel. 2022.
Imagen principal: Imagen de Freepik
Physiology, Hemostasis. Andrew LaPelusa; Heeransh D. Dave. May 8, 2022. National Library of Medicine. Recuperado de https://www.ncbi.nlm.nih.gov/books/NBK545263/